Archive
Highly Aggressive New Strain Of HIV Is Spreading Through Cuba

Given the deadly global rampage that HIV has been on for the past few decades, you’re all probably familiar with the virus. But you may not be aware that there are two types of HIV—HIV-1 and HIV-2—with the former being significantly more prevalent worldwide. The most common type of HIV-1 is then further divided into distinct subtypes, some of which are associated with a more rapid progression to AIDS. If these different viruses meet in an infected person, for example if someone infected with one subtype is exposed to a different one, they can exchange bits of their genetic material to create a new virus.
One of these so called “circulating recombinant forms” is currently spreading through Cuba, and it’s unfortunately extremely aggressive. Individuals infected with this hybrid virus, which is a mix of three different HIV-1 subtypes, progress to AIDS more than three times faster than average. Now, scientists have scrutinized this particularly pathogenic strain, which has yielded insight into the traits that have bestowed it with this deadly efficiency. The findings have been published in EBioMedicine.
Before HIV can get inside our cells, it first needs to bind to receptors on the surface called CD4. While this is an essential first step, it’s insufficient to get the virus inside. This is where anchoring points, called coreceptors, come in, which HIV also has to latch onto to gain entry. There are two coreceptors, CCR5 and CXCR4, and around 90% of newly transmitted HIV uses the former.
CXCR4-using viruses emerge in around 50% of individuals, but this usually takes around five years from infection. These viruses are associated with a more pronounced depletion of immune cells, but whether this shift in coreceptor preference is a cause or consequence of disease progression is unknown. Interestingly, however, the aggressive recombinant currently spreading through Cuba starts to use CXCR4 very early on in infection, and researchers think this is likely contributing to the observed rapid progression to AIDS.
To find this out, researchers examined 73 recently infected patients in Cuba, 52 who had rapidly progressed to AIDS within three years and 21 without AIDS. Then, they compared the blood of these individuals with 22 patients who had progressed to AIDS after the period typically expected, which is around 10-15 years without treatment.
They found that all those who had progressed to AIDS within three years of infection were infected with a recombinant called CRF19, which is a mixture of subtypes A, D and G. Interestingly, infection with A/D recombinants has previously been reported to result in rapid progression to AIDS, but no CRFs had been exclusively associated with rapid progression. Furthermore, those infected with CRF19 had abnormally high levels of an immune response molecule called RANTES, which acts by binding to CCR5. Without this coreceptor available for binding, CRF19 may have been forced to bypass that anchor point and go straight for CXCR4. Since the switch to CXCR4 usage is associated with progression to AIDS, this could explain why those infected with CRF19 developed AIDS so early on.
Another reason that CRF19 might be so pathogenic is that it has an enzyme, called protease, from subtype D, which is known to be very efficient. This enzyme helps the virus form mature particles, which is an essential stage in the virus life cycle.
Via KU Leuven and EBioMedicine
Team prevents memory problems caused by sleep deprivation

The hippocampus of a mouse in the University of Pennsylvania study glows green where cells have taken up a receptor that triggers a cAMP signalling pathway. After administering the ligand to the receptor, researchers could selectively boost cAMP levels in this region and this cell type only. They found that ‘rescuing’ these cells with a shot of cAMP preventing the memory problems that sleep loss can induce. Credit: University of Pennsylvania
Sleep is a critical period for memory consolidation, and most people don’t get enough. Research has shown that even brief periods of sleep deprivation can lead to deficits in memory formation.
In a new study, published in the Journal of Neuroscience, a team led by scientists from the University of Pennsylvania found that a particular set of cells in a small region of the brain are responsible for memory problems after sleep loss. By selectively increasing levels of a signaling molecule in these cells, the researchers prevented mice from having memory deficits.
Robbert Havekes was the lead author on the study. He is a research associate in the lab of Ted Abel, the study’s senior author and Brush Family Professor of Biology in Penn’s School of Arts & Sciences. Coauthors from the Abel lab included Jennifer C. Tudor and Sarah L. Ferri. They collaborated with Arnd Baumann of Forschungszentrum Jülich, Germany, and Vibeke M. Bruinenberg and Peter Meerlo of the University of Groningen, The Netherlands.
In 2009, a group from Abel’s lab published a study in Nature that identified the cyclic AMP, or cAMP, signaling pathway as playing a role in sleep-loss-associated memory problems. Whereas depriving mice of sleep impaired their spatial memory, restoring levels of cAMP in their brain prevented this effect.
“The challenge following this important study,” Abel said, “was to determine if the impact of sleep deprivation was mediated by particular regions of the brain and particular neural circuits. We suspected that the hippocampus, the brain region that mediates spatial navigation and contextual memory, was critical.”
In the current work, they set out to answer these questions. They targeted excitatory neurons because of their importance in transmitting signals in the brain and the fact that their functioning relies on cAMP signaling. The limitation of previous studies was that they lacked a way to increase cAMP in just one area of the brain in a cell-type specific fashion. Havekes, Abel and colleagues devised a way of doing this that they term a “pharmacogenetic” approach, blending genetic modification and drug administration.
They engineered a non-pathogenic virus to harbor the gene encoding the receptor for the protein octopamine, which triggers cAMP pathway activation in fruit flies but is not naturally found in the brains of mice. The researchers injected this virus into the hippocampus of mice so that the excitatory neurons in that region alone would express the octopamine receptor.
“It sounds weird. Why would you put a receptor there that is never going to be activated?” Havekes said. “The trick is, you follow that up by giving mice the ligand of the receptor, which is octopamine, and that will activate the receptors only where they are present.”
The team confirmed that only the excitatory hippocampal neurons expressed the receptor and that they could selectively increase cAMP levels in only these cells by giving the mice a systemic injection of octopamine.
“This way, we could manipulate the cAMP pathways that we previously saw being affected by sleep deprivation but selectively in specific neural circuits in the brain,” Havekes says.
With this pharmacogenetic tool in hand, Havekes, Abel and colleagues began the sleep deprivation tests with the mice expressing the octopamine receptor in their hippocampus. First the researchers trained mice in a spatial memory task. They put them in a box that had three different objects, each in a distinct location.
Then, because previous research had shown that cAMP signaling contributes to hippocampus-dependent memory consolidation in two time windows—first directly after training and again three to four hours after training—the researchers gave mice in the experimental groups injections of octopamine in both of these windows to boost cAMP levels.
Mice receiving the cAMP boost were divided into two groups: One was left to sleep undisturbed, while the other was sleep-deprived for five hours by gently tapping their cage or rearranging their bedding.
One full day after the initial training, all of the mice were tested again. This time, there was a twist: one of the objects originally in the box had been moved to a new location.
“If the mice had learned and remembered the location of the objects during their training, then they would realize, okay, this is the object that has moved, and they’ll spend more time exploring that particular object,” Havekes explained. “If they didn’t remember well, they would explore all the objects in a random fashion.”
The researchers found that the sleep-deprived mice that received the octopamine injections spent more time exploring the object that had moved, just as mice that had not been sleep deprived did. On the other hand, sleep-deprived mice that didn’t express the receptor explored all the objects at random, a sign that they had failed to remember the locations of the objects from their initial training as a result of the brief period of sleep deprivation.
“What we’ve shown is this memory loss due to sleep deprivation is really dependent on misregulation of cAMP signaling in the excitatory neurons of the hippocampus,” Havekes said.
As a next step, the group would like to explore what cAMP is doing to help consolidate memory. They would also like to investigate how other cell types in the brain, such as astrocytes, might be affected. And finally, while this study focused on the impact of a brief period of sleep deprivation, Havekes is curious to know how not getting enough sleep on a daily basis, as is more similar to human experiences, might be affecting memory.
“Thinking about people who do shift work or doctors who work long hours, if we can tackle the cognitive problems that result from sleep loss, that would be a great thing,” Havekes said.
“At least in the mouse using these sophisticated tools, we’re able to reverse the negative impact of sleep deprivation on cognition,” Abel said.
A Flu Virus That Killed Millions In 1918 Has Now Been Recreated

Spanish flu
Scientists have recreated a nearly exact replicate of the deadly flu virus that killed an estimated 50 million in the 1918 Spanish flu pandemic.
But don’t worry, they say it’s totally safe.
Researchers at the University of Wisconsin-Madison reverse engineered an influenza virus from a similar one found in birds, combining several strains to create one that is nearly identical to the one that caused the 1918 outbreak. They then mutated the genes to make it airborne, and to study how it spreads between animals.
“Our research indicates the risks inherent in circulating avian influenza viruses,” Yoshihiro Kawaoka, the scientist who led the research team, told VICE News. “Continued surveillance of avian influenza viruses — and not only viruses that we know pose risks for humans, such as H5N1 and H7N9 influenza viruses, and attention to pandemic preparedness measures is important.”
According to the statement summarizing the project published this week, the “analyses revealed the global prevalence of avian influenza virus genes whose proteins differ only a few amino acids from the 1918 pandemic influenza virus, suggesting that 1918-like pandemic viruses may emerge in the future.”
In other words, a common avian flu virus that has been circulating in wild ducks is pretty much the exact same one that infected humans a century ago. And now is in a lab.
The research was funded by the National Institute of Health as a way to find out more about similar virus’ and their transmissibility from animals to humans. It was done in a lab that complied with full safety and security regulations, said Carole Heilman, director of the Division of Microbiology and Infectious Diseases, at National Institute of Allergy and Infectious Diseases (NIAD), a division of NIH.
“It was an question of risk versus benefit,” Heilman told VICE News. “We determined that the risk benefit ratio was adequate if we had this type of safety regulations.”
But many scientists disagree and have condemned research that recreates virus’ such this, stating that if released accidentally, a virus could spread to humans and cause a pandemic. Marc Lipsitch, an epidemiologist at Harvard, has criticized research such as Kawaoka’s as unnecessarily risky.
“There is a quantifiable possibility that these novel pathogens could be accidentally or deliberately released. Exacerbating the immunological vulnerability of human populations to PPPs is the potential for rapid global dissemination via ever-increasing human mobility,” Lipsitch said in a paper about experiments with transmissible virus’. “The dangers are not just hypothetical.”
Lipsitch points out that many of the H1N1 flu outbreaks that have occurred between 1977 and 2009 were a result of a lab accident.
Kawaoka disagrees, saying, “We maintain that it is better to know as much as possible about the risk posed by these viruses so we may be able to identify the risk when viruses with pandemic potential emerge, and have effective countermeasures on-hand or ready for development.”
This article originally appeared at VICE News. Check them out on YouTube, Facebook, and Instagram. Copyright 2014. Follow VICE News on Twitter.
Pithovirus: 30,000-year-old giant virus ‘comes back to life’
A new virus called Pithovirus sibericum has been isolated from 30,000 year old Siberian permafrost. It is the oldest DNA virus of eukaryotes ever isolated, showing that viruses can retain infectivity in nature for very long periods of time.
Pithovirus was isolated by inoculating cultures of the amoeba Acanthamoeba castellani with samples taken in the year 2000 from 30 meters below the surface of a late Pleistocene sediment in the Kolyma lowland region. This amoeba had been previously used to propagate other giant viruses, such as Mimivirus and Pandoravirus. Light microscopy of the cultures revealed the presence of ovoid particles which were subsequently shown by electron microscopy to resemble those of Pandoravirus. Pithovirus particles are flask-shaped and slightly larger than Pandoravirus – 1.5 microns long, 500 nm in diameter, encased by a 60 nm thick membrane. One end of the virus particle appears to be sealed with what the authors call a cork (photo). This feature, along with the shape of the virus particle, inspired the authors to name the new isolate Pithovirus, from the Greek word pithos which refers to the amphora given to Pandora. The name therefore refers both to the morphology of the virus particle and its similarity to Pandoravirus.
Although the Pithovirus particle is larger than Pandoravirus, the viral genome – which is a double-stranded molecule of DNA – is smaller, a ‘mere 610,033 base pairs’, to use the authors’ words (the Pandoravirus genome is 2.8 million base pairs in length). There are other viruses with genomes of this size packed into much smaller particles – so why is the Pithovirus particle so large? Might it have recently lost a good deal of its genome and the particle size has not yet caught up? One theory of the origin of viruses is that they originated from cells and then lost genes on their way to becoming parasitic.
We now know of viruses from two different families that have similar morphology: an amphora-like shape, an apex, and a thick electron-dense tegument covered by a lipid membrane enclosing an internal compartment. This finding should not be surprising: similar viral architectures are known to span families. The icosahedral architecture for building a particle, for example, can be found in highly diverse viral families. The question is how many viruses are built with the pithovirus/pandoravirus structure. Prof. Racaniello’s guess would be many, and they could contain either DNA genomes. We just need to look for them, a process, as the authors say that ‘will remain a challenging and serendipitous process’.
Despite the physical similarity with Pandoravirus, the Pithovirus genome sequence reveals that it is barely related to that virus, but more closely resembles members of the Marseillviridae, Megaviridae, and Iridoviridae. These families all contain large icosahedral viruses with DNA genomes. Only 32% of the 467 predicted Pithovirus proteins have homologs in protein databases (this number was 61% for Mimivirus and 16% for Pandoravirus). In contrast to other giant DNA viruses, the genome of Pithovirus does not encode any component of the protein synthesis machinery. However the viral genome does encode the complete machinery needed to produce mRNAs. These proteins are present in the purified Pithovirus particle. Pithovirus therefore undergoes its entire replication cycle in the cytoplasm, much like other large DNA viruses such as poxviruses.
Pithovirus is an amazing virus that hints about the yet undiscovered viral diversity that awaits discovery. Its preservation in a permafrost layer suggests that these regions might harbor a vast array of infectious organisms that could be released as these regions thaw or are subjected to exploration for mineral and oil recovery. A detailed analysis of the microbes present in these regions is clearly needed, both by the culture technique used in this paper and by metagenomic analysis, to assess whether any constitute a threat to animals.
The above story is reprinted from materials provided by Virology blog: About Viruses and Viral Disease.
New virus isolated from patients with severe brain infections
Researchers have identified a new virus in patients with severe brain infections in Vietnam. Further research is needed to determine whether the virus is responsible for the symptoms of disease.
The virus was found in a total of 28 out of 644 patients with severe brain infections in the study, corresponding to around 4%, but not in any of the 122 patients with non-infectious brain disorders that were tested.
Infections of the brain and central nervous system are often fatal and patients who do survive, often young children and young adults, are left severely disabled. Brain infections can be caused by a range of bacterial, parasitic, fungal and viral agents, however, doctors fail to find the cause of the infection in more than half of cases despite extensive diagnostic efforts. Not knowing the causes of these brain infections makes public health and treatment interventions impossible.
Researchers at the Oxford University Clinical Research Unit, Wellcome Trust South East Asia Major Overseas Programme and the Academic Medical Center, University of Amsterdam identified the virus, tentatively named CyCV-VN, in the fluid around the brain of two patients with brain infections of unknown cause. The virus was subsequently detected in an additional 26 out of 642 patients with brain infections of known and unknown causes.
Using next-generation gene sequencing techniques, the team sequenced the entire genetic material of the virus, confirming that it represents a new species that has not been isolated before. They found that it belongs to a family of viruses called the Circoviridae, which have previously only been associated with disease in animals, including birds and pigs.
Dr Rogier van Doorn, Head of Emerging Infections at the Wellcome Trust Vietnam Research Programme and Oxford University Clinical Research Unit Hospital for Tropical Diseases in Vietnam, explains: “We don’t yet know whether this virus is responsible for causing the serious brain infections we see in these patients, but finding an infectious agent like this in a normally sterile environment like the fluid around the brain is extremely important. We need to understand the potential threat of this virus to human and animal health.”
The researchers were not able to detect CyCV-VN in blood samples from the patients but it was present in 8 out of 188 fecal samples from healthy children. The virus was also detected in more than half of fecal samples from chickens and pigs taken from the local area of one of the patients from whom the virus was initially isolated, which may suggest an animal source of infection.
Dr Le Van Tan, Oxford University Clinical Research Unit, Wellcome Trust Major Overseas Programme, said: “The evidence so far seems to suggest that CyCV-VN may have crossed into humans from animals, another example of a potential zoonotic infection. However, detecting the virus in human samples is not in itself sufficient evidence to prove that the virus is causing disease, particularly since the virus could also be detected in patients with other known viral or bacterial causes of brain infection. While detection of this virus in the fluid around the brain is certainly remarkable, it could still be that it doesn’t cause any harm. Clearly we need to do more work to understand the role this virus may play in these severe infections.”
The researchers are currently trying to grow the virus in the laboratory using cell culture techniques in order to develop a blood assay to test for antibody responses in patient samples, which would indicate that the patients had mounted an immune response against the virus. Such a test could also be used to study how many people in the population have been exposed to CyCV-VN without showing symptoms of disease.
The team are collaborating with scientists across South East Asia and in the Netherlands to determine whether CyCV-VN can be detected in patient samples from other countries and better understand its geographical distribution.
Professor Menno de Jong, head of the Department of Medical Microbiology of the Academic Medical Centre in Amsterdam said: “Our research shows the importance of continuing efforts to find novel causes of important infectious diseases and the strength of current technology in aid of these efforts.”
Journal reference: L.V. Tan et al . Identification of a new cyclovirus in cerebrospinal fluid of patients with acute central nervous system infections. mBio, June 2013. DOI: 10.1128/mBio.00231-13
The above story is reprinted from materials provided by Wellcome Trust, via MedicalXpress.
How Herpesvirus Invades Nervous System
Northwestern Medicine scientists have identified a component of the herpesvirus that “hijacks” machinery inside human cells, allowing the virus to rapidly and successfully invade the nervous system upon initial exposure.
Led by Gregory Smith, associate professor in immunology and microbiology at Northwestern University Feinberg School of Medicine, researchers found that viral protein 1-2, or VP1/2, allows the herpesvirus to interact with cellular motors, known as dynein. Once the protein has overtaken this motor, the virus can speed along intercellular highways, or microtubules, to move unobstructed from the tips of nerves in skin to the nuclei of neurons within the nervous system.
This is the first time researchers have shown a viral protein directly engaging and subverting the cellular motor; most other viruses passively hitch a ride into the nervous system.
“This protein not only grabs the wheel, it steps on the gas,” says Smith. “Overtaking the cellular motor to invade the nervous system is a complicated accomplishment that most viruses are incapable of achieving. Yet the herpesvirus uses one protein, no others required, to transport its genetic information over long distances without stopping.”
Herpesvirus is widespread in humans and affects more than 90 percent of adults in the United States. It is associated with several types of recurring diseases, including cold sores, genital herpes, chicken pox, and shingles. The virus can live dormant in humans for a lifetime, and most infected people do not know they are disease carriers. The virus can occasionally turn deadly, resulting in encephalitis in some.
Until now, scientists knew that herpesviruses travel quickly to reach neurons located deep inside the body, but the mechanism by which they advance remained a mystery.
Smith’s team conducted a variety of experiments with VP1/2 to demonstrate its important role in transporting the virus, including artificial activation and genetic mutation of the protein. The team studied the herpesvirus in animals, and also in human and animal cells in culture under high-resolution microscopy. In one experiment, scientists mutated the virus with a slower form of the protein dyed red, and raced it against a healthy virus dyed green. They observed that the healthy virus outran the mutated version down nerves to the neuron body to insert DNA and establish infection.
“Remarkably, this viral protein can be artificially activated, and in these conditions it zips around within cells in the absence of any virus. It is striking to watch,” Smith says.
He says that understanding how the viruses move within people, especially from the skin to the nervous system, can help better prevent the virus from spreading.
Additionally, Smith says, “By learning how the virus infects our nervous system, we can mimic this process to treat unrelated neurologic diseases. Even now, laboratories are working on how to use herpesviruses to deliver genes into the nervous system and kill cancer cells.”
Smith’s team will next work to better understand how the protein functions. He notes that many researchers use viruses to learn how neurons are connected to the brain.
“Some of our mutants will advance brain mapping studies by resolving these connections more clearly than was previously possible,” he says.
Story Source:
The above story is reprinted from materials provided by Northwestern University, via EurekAlert!, a service of AAAS.
Journal Reference:
Sofia V. Zaichick, Kevin P. Bohannon, Ami Hughes, Patricia J. Sollars, Gary E. Pickard, Gregory A. Smith. The Herpesvirus VP1/2 Protein Is an Effector of Dynein-Mediated Capsid Transport and Neuroinvasion. Cell Host & Microbe, 2013; 13 (2): 193 DOI: 10.1016/j.chom.2013.01.009
Nanoparticles Laced With Bee Venom Selectively Destroy HIV Virus
Nanoparticles carrying a toxin found in bee venom can destroy human immunodeficiency virus (HIV) while leaving surrounding cells unharmed, researchers at Washington University School of Medicine in St. Louis have shown. The finding is an important step toward developing a vaginal gel that may prevent the spread of HIV, the virus that causes AIDS.
“Our hope is that in places where HIV is running rampant, people could use this gel as a preventive measure to stop the initial infection,” says Joshua L. Hood, MD, PhD, a research instructor in medicine.
The study appears in the current issue of Antiviral Therapy.
Bee venom contains a potent toxin called melittin that can poke holes in the protective envelope that surrounds HIV, and other viruses. Large amounts of free melittin can cause a lot of damage. Indeed, in addition to anti-viral therapy, the paper’s senior author, Samuel A. Wickline, MD, the J. Russell Hornsby Professor of Biomedical Sciences, has shown melittin-loaded nanoparticles to be effective in killing tumor cells.
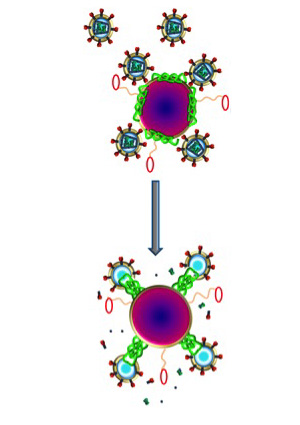
Nanoparticles (purple) carrying melittin (green) fuse with HIV (small circles with spiked outer ring), destroying the virus’s protective envelope. Molecular bumpers (small red ovals) prevent the nanoparticles from harming the body’s normal cells, which are much larger in size.
The new study shows that melittin loaded onto these nanoparticles does not harm normal cells. That’s because Hood added protective bumpers to the nanoparticle surface. When the nanoparticles come into contact with normal cells, which are much larger in size, the particles simply bounce off. HIV, on the other hand, is even smaller than the nanoparticle, so HIV fits between the bumpers and makes contact with the surface of the nanoparticle, where the bee toxin awaits.
“Melittin on the nanoparticles fuses with the viral envelope,” Hood says. “The melittin forms little pore-like attack complexes and ruptures the envelope, stripping it off the virus.”
According to Hood, an advantage of this approach is that the nanoparticle attacks an essential part of the virus’ structure. In contrast, most anti-HIV drugs inhibit the virus’s ability to replicate. But this anti-replication strategy does nothing to stop initial infection, and some strains of the virus have found ways around these drugs and reproduce anyway.
“We are attacking an inherent physical property of HIV,” Hood says. “Theoretically, there isn’t any way for the virus to adapt to that. The virus has to have a protective coat, a double-layered membrane that covers the virus.”
Beyond prevention in the form of a vaginal gel, Hood also sees potential for using nanoparticles with melittin as therapy for existing HIV infections, especially those that are drug-resistant. The nanoparticles could be injected intravenously and, in theory, would be able to clear HIV from the blood stream.
“The basic particle that we are using in these experiments was developed many years ago as an artificial blood product,” Hood says. “It didn’t work very well for delivering oxygen, but it circulates safely in the body and gives us a nice platform that we can adapt to fight different kinds of infections.”
Since melittin attacks double-layered membranes indiscriminately, this concept is not limited to HIV. Many viruses, including hepatitis B and C, rely on the same kind of protective envelope and would be vulnerable to melittin-loaded nanoparticles.
While this particular paper does not address contraception, Hood says the gel easily could be adapted to target sperm as well as HIV. But in some cases people may only want the HIV protection.
“We also are looking at this for couples where only one of the partners has HIV, and they want to have a baby,” Hood says. “These particles by themselves are actually very safe for sperm, for the same reason they are safe for vaginal cells.”
While this work was done in cells in a laboratory environment, Hood and his colleagues say the nanoparticles are easy to manufacture in large enough quantities to supply them for future clinical trials.
Journal referrence:
Hood JL, Jallouck AP, Campbell N, Ratner L, Wickline SA. Cytolytic nanoparticles attenuate HIV-1 infectivity. Antiviral Therapy. Vol. 19: 95 – 103. 2013
Source:
Innate immune system can kill HIV when a viral gene is deactivated
Human cells have an intrinsic capacity to destroy HIV. However, the virus has evolved to contain a gene that blocks this ability. When this gene is removed from the virus, the innate human immune system destroys HIV by mutating it to the point where it can no longer survive.
This phenomenon has been shown in test tube laboratory experiments, but now researchers at the University of North Carolina School of Medicine have demonstrated that the same phenomenon occurs in a humanized mouse model, suggesting a promising new target for tackling the virus, which has killed nearly 30 million people worldwide since it first appeared three decades ago.
A family of human proteins called APOBEC3 effectively restrict the growth of HIV and other viruses, but this action is fully counteracted by the viral infectivity factor gene (vif) in HIV. In the study, researchers intravenously infected humanized mice with HIV. They found that the most commonly transmitted strains of HIV are completely neutralized by APOBEC3 proteins when vif is removed from the virus.
“Without the vif gene, HIV can be completely destroyed by the body’s own immune system,” said J. Victor Garcia, PhD, professor of medicine at the UNC School of Medicine and senior author on the study. “These results suggest a new target for developing drugs fully capable of killing the virus.”
Garcia and his colleagues pioneered the humanized mouse model used for these studies. The aptly named “BLT” mouse is created by introducing human bone marrow, liver and thymus tissues into animals without an immune system of their own. The mice have a fully functioning human immune system and can be infected with HIV in the same manner as humans. In previous research, Garcia and his team have effectively prevented intravenous, rectal, vaginal and oral transmission of HIV in the mice with pre-exposure prophylaxis (PrEP).
For the current study, Garcia and his colleagues also infected BLT mice with another, highly harmful strain of the virus. The results show that this strain of HIV does continue to replicate, even without vif, but at a much slower rate and without harming the human immune system. Further, the researchers found that virus replication in this case was limited to one tissue—the thymus—in the entire body.
“These findings demonstrate a fundamental weakness in HIV,” said John F. Krisko, PhD, lead author on the study. “If this weakness can be exploited, it might eventually lead to a cure for HIV/AIDS,” Krisko said.
Journal reference:
John F. Krisko, Francisco Martinez-Torres, John L. Foster, J. Victor Garcia. HIV Restriction by APOBEC3 in Humanized Mice. PLoS Pathogens, 2013; 9 (3): e1003242 DOI: 10.1371/journal.ppat.1003242
Provided by University of North Carolina Health Care
Source 3: Protein Essential for Ebola Virus Infection Is a Promising Antiviral Target
In separate papers published online in Nature, two research teams report identifying a critical protein that Ebola virus exploits to cause deadly infections. The protein target is an essential element through which the virus enters living cells to cause disease.
The first study was led by four senior scientists: Sean Whelan, associate professor of microbiology and immunobiology at Harvard Medical School; Kartik Chandran, assistant professor at Albert Einstein College of Medicine; John Dye at the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID) and Thijn Brummelkamp, originally at the Whitehead Institute for Biomedical Research and now at the Netherlands Cancer Institute. The second study was led by James Cunningham, a Harvard Medical School associate professor of medicine at Brigham and Women’s Hospital, and also co-authored by Chandran.
“This research identifies a critical cellular protein that the Ebola virus needs to cause infection and disease,” explained Whelan, who is also co-director of the HMS Program in Virology. “The discovery also improves chances that drugs can be developed that directly combat Ebola infections.”
Both papers are published in the August 24 online issue of Nature.
The African Ebola virus — and its cousin, Marburg virus — are known as the filoviruses. Widely considered one of the most dangerous infections known, Ebola was first identified in 1976 in Africa near the Ebola River, an area in Sudan and the Democratic Republic of the Congo. Infections cause severe hemorrhage, multiple organ failure and death. No one quite knows how the virus is spread, and there are no available vaccines or anti-viral drugs that can fight the infections.
Through conducting a genome-wide genetic screen in human cells aimed at identifying molecules essential for Ebola’s virulence, Whelan and his colleagues homed in on Niemann-Pick C1 (NPC1).
NPC1 has been well known in the biomedical literature. Primarily associated with cholesterol metabolism, this protein, when mutated, causes a rare genetic disorder in children, Niemann-Pick disease.
Using cells derived from these patients, the group found that this mutant form of NPC1 also completely blocks infection by the Ebola virus. They also demonstrated that mice carrying a mutation in the NPC1 gene resisted Ebola infection. Similar resistance was found in cultured cells in which the normal molecular structure of the Niemann-Pick protein has been altered.
In other words, targeting NPC1 has real therapeutic potential. While such a treatment may also block the cholesterol transport pathway, short-term treatment would likely be tolerated.
Indeed in the accompanying paper, Cunningham’s group describes such a potential inhibitor.
Cunningham and his group at Brigham and Women’s Hospital investigated Ebola by using a robotic method developed by their colleagues at the National Small Molecule Screening Laboratory at Harvard Medical School to screen tens of thousands of compounds. The team identified a novel small molecule that inhibits Ebola virus entry into cells by more than 99 percent.
The team then used the inhibitor as a probe to investigate the Ebola infection pathway and found that the inhibitor targeted NPC1.
For Cunningham and Chandran, this finding builds on a 2005 paper of theirs for which Whelan was also a collaborator. In that study, he and his group discovered how Ebola exploits a protein called cathepsin B. This new study completes the puzzle. It now seems that cathepsin B interacts with Ebola in a way that preps it to subsequently bind with NPC1.
“It is interesting that NPC1 is critical for the uptake of cholesterol into cells, which is an indication of how the virus exploits normal cell processes to grow and spread,” said Cunningham. “Small molecules that target NPC1 and inhibit Ebola virus infection have the potential to be developed into anti-viral drugs.”
The paper coauthored by Whelan was funded by the U.S. National Institute of Allergy and Infectious Diseases and the National Human Genome Research Institute, the U.S. Army, and the Burroughs Wellcome Foundation. Cunningham’s work was funded by the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases at Harvard Medical School.
Story Source:
The above story is reprinted from materials provided by Harvard Medical School, via ScienceDaily. The original article was written by Robert Cooke and Lori Shanks.
Journal References:
- Jan E. Carette, Matthijs Raaben, Anthony C. Wong, Andrew S. Herbert, Gregor Obernosterer, Nirupama Mulherkar, Ana I. Kuehne, Philip J. Kranzusch, April M. Griffin, Gordon Ruthel, Paola Dal Cin, John M. Dye, Sean P. Whelan, Kartik Chandran, Thijn R. Brummelkamp. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature, 2011; DOI: 10.1038/nature10348
- Marceline Côté, John Misasi, Tao Ren, Anna Bruchez, Kyungae Lee, Claire Marie Filone, Lisa Hensley, Qi Li, Daniel Ory, Kartik Chandran, James Cunningham. Small molecule inhibitors reveal Niemann–Pick C1 is essential for Ebola virus infection. Nature, 2011; DOI: 10.1038/nature10380
Source 2: Scientists Identify Point of Entry for Deadly Ebola Virus
Ebola virus, the cause of Ebola hemorrhagic fever (EHF), is one of the deadliest known viruses affecting humans. Like anthrax and smallpox virus, Ebola virus is classified by the U.S. Centers for Disease Control and Prevention (CDC) as a category A bioterrorism agent. Currently, there is no vaccine to prevent EHF, and patients are treated only for their symptoms.Although outbreaks are rare, Ebola virus, the cause of Ebola hemorrhagic fever (EHF), is one of the deadliest known viruses affecting humans. According to the World Health Organization (WHO), approximately 1,850 EHF cases with more than 1,200 deaths have been documented since the virus was identified in 1976.

This negatively-stained transmission electron micrograph (TEM) revealed some of the ultrastructural curvilinear morphologic features displayed by the Ebola virus discovered from the Ivory Coast of Africa. (Credit: Charles Humphrey). (up)
EHF’s clinical presentation can be devastating: fever, intense weakness, and joint and muscle aches progress to diarrhea, vomiting, and in some cases, internal and external bleeding caused by disintegrating blood vessels. Currently, there is no approved vaccine and patients are treated only for their symptoms. Like anthrax and smallpox virus, Ebola virus is classified as a category A bioterrorism agent by the U.S. Centers for Disease Control and Prevention (CDC).
Until now, however, researchers had only a limited understanding of how Ebola virus gains entry to a host cell.
Using an unusual human cell line, Whitehead Institute scientists and collaborators from Harvard Medical School, Albert Einstein College of Medicine and U.S. Army Medical Research Institute of Infectious Diseases, have identified the Niemann-Pick C1 (NPC1) protein as crucial for Ebola virus to enter cells and begin replicating. The discovery may offer a new and better approach for the development of antiviral therapeutics, as it would target a structure in the host cell rather than a viral component.
The findings are reported online in Nature this week.
Where all of us inherit one copy of each chromosome from each of our two parents, cell lines exist with only a single set, and thus with a single copy of each individual gene, instead of the usual two. Using an unusual human cell line of this type, Whitehead Institute researchers and their collaborators performed a genetic screen and identified a protein used by Ebola virus to gain entry into cells and begin replicating. The discovery may offer a new approach for the development of antiviral therapeutics.
“Right now, people make therapeutics to inactivate the pathogen itself. But the problem is that pathogens can quickly change and escape detection and elimination by the immune system,” says former Whitehead Fellow Thijn Brummelkamp, now a group leader at the Netherlands Cancer Institute (NKI). “Here we get a good idea of the host genes that are needed for the pathogen to enter the cell for replication. Perhaps by generating therapeutics against those host factors, we would have a more stable target for antiviral drugs.”
The method developed by the Brummelkamp lab to identify host factors relies on gene disruption — knocking out gene function in the host cells, one gene at a time — and documenting which cells survive due to mutations that afford protection from viral entry.
But human cells are diploid with two copies of each chromosome and its genes. Researchers can reliably target and knock out one copy of a gene, but doing so for both copies is far more difficult and time-consuming. If only a single copy is silenced, the other continues to function normally and masks any effect of the knockout.
To sidestep this obstacle, Jan Carette, a first co-author on the Nature paper and a former postdoctoral researcher in the Brummelkamp lab, employed a technique he had previously applied to study the cytolethal distending toxin (CDT) family that is secreted by multiple pathogenic bacteria, including Escherichia coli, Shigella dysenteriae, and Haemophilus ducreyi. Each bacterial species has developed its own twists on the CDT structure, which may link to the target tissues of the toxin’s bacterium.
In his CDT work published in Nature Biotechnology, Carette together with co-lead authors of Whitehead Member Hidde Ploegh’s lab, used a line of haploid cells isolated from a chronic myeloid leukemia (CML) patient. Because these cells, called KBM7 cells, have only one copy of each chromosome except chromosome 8, the researchers could disrupt the expression of each gene and screen for mutants with the desired properties, in this case survival of a lethal dose of toxin.
After knocking out individual genes by disrupting the normal structure of the gene, the resulting mutant KBM7 cells were exposed to various CDTs. In the cells that survived, Carette and coauthors knew that genes that had been disrupted were somehow crucial to CDT intoxication. By analyzing the surviving cell’s genomes, Carette and coauthors identified ten human proteins that are used by CDTs during intoxication, and those host factors seem to be tailored to each CDT’s targeted cell.
“I found it surprising that there is quite some specificity in the entry routes for each toxin,” says Carette. “If you take CDTs that are very similar to each other in structure, you could still see significant differences in the host factors they require to do their job. So it seems that every pathogen evolved a specific and unique way of its toxin entering the cells.”
To study Ebola virus, Carette and co-lead authors from Harvard Medical School and the Albert Einstein College of Medicine made use of an otherwise harmless virus cloaked in the Ebola virus glycoprotein coat. Using this virus and by altering the haploid cells somewhat, Carette and coauthors were able to pinpoint the cellular genes that Ebola virus relies on to enter the cell.
Carette and coauthors identified as necessary for Ebola virus entry several genes involved in organelles that transport and recycle proteins. One gene in particular stood out, NPC1, which codes for a cholesterol transport protein, and is necessary for the virus to enter the cell’s cytoplasm for replication. Mutations in this gene cause a form of Niemann-Pick disease, an ultimately fatal neurological disorder diagnosed mainly in children.
Collaborators at the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID) tested the effects of active Ebola virus on mice that had one copy of the NPC1 gene knocked out. Control mice, with two functioning copies of the NPC1 gene, quickly succumbed to infection, while the NPC1 knockout mice were largely protected from the virus.
“This is pretty unexpected,” says Carette, who is currently an Acting Assistant Professor in Microbiology & Immunology at Stanford School of Medicine. “This might imply that genetic mutations in the NPC1 gene in humans could make some people resistant to this very deadly virus. And now that we know that NPC1 is an Ebola virus host factor, it provides a strong platform from which to start developing new antivirals.”
This research was supported by the National Institutes of Health (NIH), the U.S. Army, Boehringer Ingelheim Fonds and a Burroughs Wellcome Award.
Story Source:
The above story is reprinted from materials provided by Whitehead Institute for Biomedical Research, via ScienceDaily. The original article was written by Nicole Giese.
Journal References:
- Jan E. Carette, Matthijs Raaben, Anthony C. Wong, Andrew S. Herbert, Gregor Obernosterer, Nirupama Mulherkar, Ana I. Kuehne, Philip J. Kranzusch, April M. Griffin, Gordon Ruthel, Paola Dal Cin, John M. Dye, Sean P. Whelan, Kartik Chandran, Thijn R. Brummelkamp. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature, 2011; DOI: 10.1038/nature10348
- Jan E Carette, Carla P Guimaraes, Irene Wuethrich, Vincent A Blomen, Malini Varadarajan, Chong Sun, George Bell, Bingbing Yuan, Markus K Muellner, Sebastian M Nijman, Hidde L Ploegh, Thijn R Brummelkamp. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nature Biotechnology, 2011; 29 (6): 542 DOI: 10.1038/nbt.1857

Scribd
![]() is where my documents live!
is where my documents live!

{kind=link}